
Research Interests
I. METAL-INSULATOR TRANSITIONS
II. GEOMETRICALLY FRUSTRATED MAGNETIC MATERIALS
III. SEARCH FOR NEW MATERIALS
|
|
|
|
Research Interests
|
|
|
Metal-Insulator Transitions in Transition-Metal Oxides
|
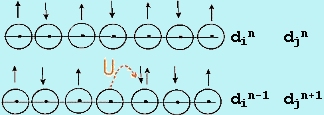
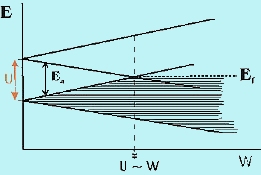
 ransition metal oxides (TMOs) have long presented a challenge to theories of electron behavior in the solid state. Since the d-levels in TMOs are partially filled, straightforward application of the molecular orbital theory to the solid state (called the band theory) always predicts electron de-localization in partially filled bands and, therefore, metallic properties. In reality, many TMOs show insulating, not metallic behavior, implying that the d-electrons are localized in discrete, atomic-like states. Mott was the first to emphasize that the short-range Coulomb repulsion of electrons can prevent formation of extended band states, thus stabilizing the localized atomic-like states. This repulsion is described in terms of a correlation energy, U, which is the energy penalty for transferring an electron between two adjacent sites (Figure 1) and is given approximately as U » IP + EA, the algebraic sum of the ionization energy and the electron affinity in the solid. The magnitude of U is a few eV or a few 102 kJ/mol. The effect of electron repulsion is to make the half-filled band insulating when the interaction between atoms is small. This situation is illustrated in Figure 2 where we compare U relative to the potential band width, W. If U > W, the d band of the transition metal is split into sub-bands and in order to transfer an electron between atoms the energy barrier due to U must be overcome and the material is insulating. Such materials are called Mott-Hubbard (MH) insulators. At the point where W ~ U, the bands overlap. Beyond this point, there is no energy gap and the solid is metallic. The study of the transition from the MH insulating state to the metallic state, called the metal/insulator transition (MIT), is a topic of great current interest. ransition metal oxides (TMOs) have long presented a challenge to theories of electron behavior in the solid state. Since the d-levels in TMOs are partially filled, straightforward application of the molecular orbital theory to the solid state (called the band theory) always predicts electron de-localization in partially filled bands and, therefore, metallic properties. In reality, many TMOs show insulating, not metallic behavior, implying that the d-electrons are localized in discrete, atomic-like states. Mott was the first to emphasize that the short-range Coulomb repulsion of electrons can prevent formation of extended band states, thus stabilizing the localized atomic-like states. This repulsion is described in terms of a correlation energy, U, which is the energy penalty for transferring an electron between two adjacent sites (Figure 1) and is given approximately as U » IP + EA, the algebraic sum of the ionization energy and the electron affinity in the solid. The magnitude of U is a few eV or a few 102 kJ/mol. The effect of electron repulsion is to make the half-filled band insulating when the interaction between atoms is small. This situation is illustrated in Figure 2 where we compare U relative to the potential band width, W. If U > W, the d band of the transition metal is split into sub-bands and in order to transfer an electron between atoms the energy barrier due to U must be overcome and the material is insulating. Such materials are called Mott-Hubbard (MH) insulators. At the point where W ~ U, the bands overlap. Beyond this point, there is no energy gap and the solid is metallic. The study of the transition from the MH insulating state to the metallic state, called the metal/insulator transition (MIT), is a topic of great current interest.
|
|
|
 |
 |
| Fig-1 correlation energy U |
Fig-2 correlation energy U vs. potential band width W |
|
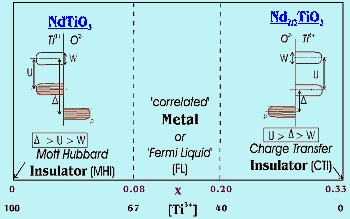
The Nd1xTiO3 system has been chosen for the study of the MIT since by introducing vacancies (x) on the rare earth sites, the degree of filling of the d-band can be controlled. For x = 1/3, Nd2/3TiO3, the oxidation state for Ti is 4+, there are no d electrons and the compound is an insulator. This compound is called a Charge Transfer Insulator (CTI) as the activation energy for conduction is now the gap between the p-band on the O2- ligands and the empty d-band on Ti. For x = 0.0, we have Ti3+ and a MH insulator for the reasons described above. Transitions from the insulating to metallic state occur with a change of Ti3+ concentration. The system can be pushed into the metallic state either by adding electrons to Nd2/3TiO3, or adding holes (removing electrons) to NdTiO3. Therefore, two MITs are expected as illustrated on the diagram in Figure 3.
|
|
|
 |
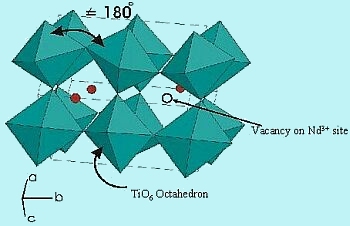
The structure type is based on an orthorhombically distorted perovskite. The space group is Pnma over the range 0.00 £ x £ 0.25. Slightly distorted TiO6 octahedra (shown in green) are corner shared while the Nd3+ ions and vacancies (red spheres) randomly occupy the interstitial sites (Figure 4). Due to the relatively small size of the Nd3+ ions, the TiOTi angle is significantly less than the ideal 180°.
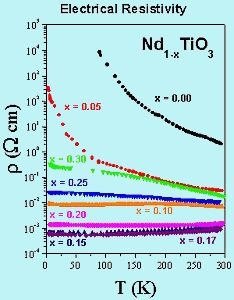
Some results of electrical resisivity measurements on several members of the Nd1-xTiO3 system are shown in Figure 5. Note that the resistivity can be varied over a range of ~ 107 simply by changing the Nd vacancy concentration. More detailed studies are currently underway. |
| Fig-3 MIT's in Nd1xTiO3 system |
|
|
|
|
 |
 |
| Fig-4 structure of Nd1xTiO3 |
Fig-5 electrical resistivity vs. temperature |
|
top of page
|
HOME | NEWS | RESEARCH | GROUP | FACILITIES | PUBLICATIONS | J. E. GREEDAN
© 2009 J.E. Greedan / McMaster University
All Rights Reserved
Last updated:
|
|


